Malattie genetiche della retina: lo stato dell’arte della cura.
- martelli.gabriella@gmail.com
- 0 Comments
Che cosa sono le malattie genetiche della retina?
Le malattie genetiche sono, in generale, le patologie che nascono per un “errore” nel nostro DNA, cioè nel nostro patrimonio genetico. Ogni cellula ha dentro di sé le “istruzioni” per costruire tutto ciò che le servirà nella vita, e quindi fare le sue funzioni. Ogni cellula conosce già il suo destino: potrà quindi specializzarsi a diventare un muscolo, uno stomaco o un rene. Queste istruzioni sono scritte in un linguaggio molto complesso (il DNA), all’interno del quale possono purtroppo capitare degli errori. Nonostante possediamo degli efficientissimi sistemi di correzione automatica, in alcuni casi gli errori nel DNA portano la cellula a crescere e svilupparsi in maniera non corretta. Questo meccanismo inceppato può portare quindi un tessuto o un organo a non funzionare adeguatamente o, addirittura, a degenerare e morire.
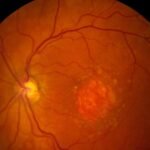
Questo è quanto accade per le malattie genetiche della retina, in cui alcune cellule che ci servono per vedere (per esempio i fotorecettori o le cellule dell’epitelio pigmentato, che nutrono la retina) non funzionano più e, progressivamente, degenerano.
Quali sono queste malattie? Quante sono?
Le malattie genetiche della retina, spesso raggruppate con il nome distrofie retiniche ereditarie, sono molte.
È difficile “contare” queste malattie poiché non c’è corrispondenza diretta tra un singolo errore del DNA (la mutazione) e una malattia. Una singola malattia, per come si presenta quando la diagnostichiamo, può essere infatti causata da diverse mutazioni. Pensate che ad oggi sono note più di 200 mutazioni che causano malattie genetiche della retina.
La più nota tra le malattie genetiche della retina è la Retinopatia pigmentosa.
Che sintomi hanno?
Le malattie genetiche della retina hanno sintomi abbastanza sovrapponibili, ed esordiscono in maniera graduale con un sintomo molto caratteristico, che è la difficoltà visiva notturna (chiamata nictalopia) o in penombra (chiamata emeralopia). Altri sintomi possono essere la riduzione del campo visivo periferico, che provoca la sensazione di vedere attraverso un “tunnel” (il paziente può inavvertitamente urtare gli oggetti intorno a sé) o la visione alterata dei colori.
I sintomi tipicamente esordiscono nella giovinezza o nel giovane adulto (15-30 anni) e, a seconda della gravità della malattia, possono progredire fino alla perdita completa della visione. I pazienti con questa malattia vivono quindi consapevolmente il dramma della perdita progressiva e graduale della vista durante la vita adulta.
Come vivono oggi questi pazienti?
La diagnosi di una patologia genetica della retina è estremamente critica, implicando uno shock psicologico per il paziente e per tutti i familiari. Cambiano le prospettive di vita, cambiano i modi in cui si affronta la realtà. Spesso la malattia deve essere cercata anche nei familiari, per capire se vi siano dei portatori sani e per capire che rischio vi sia di trasmettere la malattia alla progenie.
Sono malati giovani che per il resto sono spesso sani e attivi, che improvvisamente devono iniziare a raffrontarsi con la terribile realtà dell’ipovisione o, addirittura, della cecità. Vuol dire imparare a usare gli ausili, il Braille, le stecche, il cane guida.
Quali sono al giorno d’oggi le cure possibili per queste malattie?
Allo stato odierno la terapia “standard” di tutte queste patologie consiste nella somministrazione di integratori antiossidanti e vitamine che si limitano a ridurre i danni provocati dall’”ingranaggio bloccato” dell’errore genetico. Questi farmaci sono in grado di rallentarne leggermente la progressione, senza però impedire il destino finale della malattia, cioè l’ipovisione o la cecità.
Al giorno d’oggi vi sono 3 grandi filoni di ricerca nella cura di queste patologie:
- Le cellule staminali
- La terapia genica
- Le protesi di retina
Quali sono i principi di queste cure? Come funzionano le cellule staminali?
Le cellule staminali sono cellule teoricamente in grado di specializzarsi a diventare qualunque tessuto del nostro corpo. Sono, per semplificare molto, come le cellule di un embrione che gradualmente crescono, si sviluppano, e possono diventare (come detto prima) un rene, un cuore, un muscolo o un occhio.
La terapia con le cellule staminali, sebbene dal punto di vista squisitamente logico sembrerebbe la più vincente, è la più complessa e, al tempo stesso, rischiosa. Tralasciando gli aspetti bioetici sull’origine di queste cellule e concentrandoci su quelli clinici, l’inserimento di cellule staminali all’interno dell’occhio non garantisce purtroppo che queste riescano ad integrarsi e funzionare. La retina è un tessuto estremamente raffinato, ed è strutturato come un circuito. Non è purtroppo sufficiente che una cellula staminale, una volta inserita nel tessuto della retina, diventi, ad esempio, un fotorecettore; questa deve anche integrarsi funzionalmente nel circuito.
Il delicato equilibrio nel loro impiego deriva anche dal fatto che essere dovrebbero crescere per sostituire il tessuto malato, crescita che deve chiaramente essere limitata a tale tessuto senza diventare illimitata. Il rischio implicito che ne deriva è, infatti, dietro l’angolo.
Le cellule staminali sono tanto affascinanti quanto complesse e, sebbene ne veda il loro utilizzo nel futuro, non sono ancora realisticamente impiegabili e gli studi clinici tutt’ora in corso devono ancora raffinarsi.
E la terapia genica?
La terapia genica è invece decisamente più avanti in questo campo. Fare terapia genica significa fornire alle cellule del tessuto malato le istruzioni corrette per poter crescere e fare la loro funzione. Non significa quindi “riprogrammare” le cellule, ma dare loro solo quel frammento di DNA corretto, così da sostituirsi a quello errato e vivere senza inceppamenti del loro meccanismo. Il frammento di DNA viene consegnato alle cellule grazie ad un delicato intervento chirurgico (in cui vado ad iniettare il gene al di sotto della retina) e sfruttando un ingegnoso meccanismo di ingegneria genetica. Il principio basilare di questa cura è somministrare questi pezzetti di gene quando la cellula è ancora vitale e funzionante, cioè quando il soggetto è ancora giovane e vedente. Somministrare geni quando le cellule non sono più vitali non è purtroppo in grado di invertire il processo di degenerazione.
Presso l’IRCCS San Raffaele abbiamo avuto la fortuna di poter curare con la terapia genica un paziente, grazie al nuovo farmaco Luxturna, disegnato per curare la retinopatia pigmentosa con la specifica mutazione del gene RPE65.
Le protesi retiniche: di cosa si tratta?
Da circa 15 anni il mondo ha visto gradualmente crescere l’attività di ricerca (e l’applicazione clinica) di nuovi modelli di protesi retiniche. Si tratta di vere e proprie “retine artificiali”, di cosiddetti “microchip” per il ripristino della visione in persone cieche o gravemente ipovedenti. Sono stati progettati, e tutt’ora lo sono, diversi modelli di queste protesi,
Nel 2018, al San Raffaele di Milano, siamo stati la prima equipe chirurgica ad aver impiantato in Italia un particolare tipo di protesi, cioè il modello sotto-retinico (Alpha AMS). Abbiamo inoltre lavorato all’impianto di una protesi di ultima generazione (Nano Retina 600), costruita sull’esperienza delle precedenti protesi e che sintetizza il meglio della bioingegneria mondiale.
All’opposto della terapia genica, le protesi di retina sono esclusivamente dedicate ai pazienti nello stadio terminale della malattia, in cui sono non vedenti o severamente ipovedenti.
Le protesi retiniche sono in grado non solo di ripristinare la percezione della luce, ma anche di fornire al soggetto una discriminazione di alcuni dettagli della visione (ombre, luci, forme). Il margine di recupero ottenibile rimane comunque una visione rudimentale e troppo bassa per essere paragonata alla visione reale, motivo per cui non avrebbe alcun razionale impiantare le protesi a pazienti con residui di visione ancora fruibili. Grazie ad un intenso programma di rieducazione visiva, il paziente dovrà infatti “reimparare” a vedere, apprendendo un nuovo modo di percepire la luce.
Il principio di funzionamento si basa sulla sostituzione dei fotorecettori della retina (cioè le cellule che servono per vedere) tramite fotodiodi, dei microscopici apparati elettronici in grado di trasformare la luce in stimoli elettrici. I microchip sono grandi pochi millimetri, e vengono impiantati all’interno dell’occhio a contatto con la retina con un delicato intervento chirurgico, in modo da stimolare il circuito nervoso che naturalmente collega il cervello all’occhio. In questo modo vanno a sostituire la funzione delle cellule malate, di fatto mimando la funzione della retina non funzionante. È per altro fondamentale che il cervello abbia già avuto conoscenza del meccanismo della visione, che “si ricordi” cioè di come si fa a trasformare l’impulso elettrico in immagini, che “si ricordi di aver già visto”.
Com’è il futuro?
Non vanno fatte promesse miracolistiche, ma oneste e caute speranze di poter ripristinare una certa percezione luminosa a persone che ne sono completamente prive.
Avere una malattia rara è una condanna, fisica e psicologica. Ci si sente spesso esclusi, dimenticati. Sapere che la ricerca c’è, sapere che arriva a dei risultati e non è un “ricerca” astratta penso sia uno degli aspetti più emozionanti e avvincenti di questo percorso. Un percorso che vede camminare fianco a fianco pazienti e clinici in un viaggio comune, tra scienza, tecnologia, speranza e leale aspettativa.